

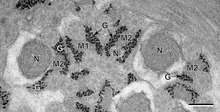
Glycogen is a multibranched polysaccharide of glucose that serves as a form of energy storage in animals,[2] fungi, and bacteria.[3] It is the main storage form of glucose in the human body.
Glycogen functions as one of three regularly used forms of energy reserves, creatine phosphate being for very short-term, glycogen being for short-term and the triglyceride stores in adipose tissue (i.e., body fat) being for long-term storage. Protein, broken down into amino acids, is seldom used as a main energy source except during starvation and glycolytic crisis (see bioenergetic systems).
In humans, glycogen is made and stored primarily in the cells of the liver and skeletal muscle.[4][5] In the liver, glycogen can make up 5–6% of the organ's fresh weight: the liver of an adult, weighing 1.5 kg, can store roughly 100–120 grams of glycogen.[4][6] In skeletal muscle, glycogen is found in a low concentration (1–2% of the muscle mass): the skeletal muscle of an adult weighing 70 kg stores roughly 400 grams of glycogen.[4] Small amounts of glycogen are also found in other tissues and cells, including the kidneys, red blood cells,[7][8][9] white blood cells,[10] and glial cells in the brain.[11] The uterus also stores glycogen during pregnancy to nourish the embryo.[12]
The amount of glycogen stored in the body mostly depends on oxidative type 1 fibres,[13][14] physical training, basal metabolic rate, and eating habits.[15] Different levels of resting muscle glycogen are reached by changing the number of glycogen particles, rather than increasing the size of existing particles[14] though most glycogen particles at rest are smaller than their theoretical maximum.[16]
Approximately 4 grams of glucose are present in the blood of humans at all times;[4] in fasting individuals, blood glucose is maintained constant at this level at the expense of glycogen stores, primarily from the liver (glycogen in skeletal muscle is mainly used as an immediate source of energy for that muscle rather than being used to maintain physiological blood glucose levels).[4] Glycogen stores in skeletal muscle serve as a form of energy storage for the muscle itself;[4] however, the breakdown of muscle glycogen impedes muscle glucose uptake from the blood, thereby increasing the amount of blood glucose available for use in other tissues.[4] Liver glycogen stores serve as a store of glucose for use throughout the body, particularly the central nervous system.[4] The human brain consumes approximately 60% of blood glucose in fasted, sedentary individuals.[4]
Glycogen is an analogue of starch, a glucose polymer that functions as energy storage in plants. It has a structure similar to amylopectin (a component of starch), but is more extensively branched and compact than starch. Both are white powders in their dry state. Glycogen is found in the form of granules in the cytosol/cytoplasm in many cell types, and plays an important role in the glucose cycle. Glycogen forms an energy reserve that can be quickly mobilized to meet a sudden need for glucose, but one that is less compact than the energy reserves of triglycerides (lipids). As such it is also found as storage reserve in many parasitic protozoa.[17][18][19]
Structure
[edit]

Glycogen is a branched biopolymer consisting of linear chains of glucose residues with an average chain length of approximately 8–12 glucose units and 2,000-60,000 residues per one molecule of glycogen.[20][21]
Like amylopectin, glucose units are linked together linearly by α(1→4) glycosidic bonds from one glucose to the next. Branches are linked to the chains from which they are branching off by α(1→6) glycosidic bonds between the first glucose of the new branch and a glucose on the stem chain.[22]
Each glycogen is essentially a ball of glucose trees, with around 12 layers, centered on a glycogenin protein, with three kinds of glucose chains: A, B, and C. There is only one C-chain, attached to the glycogenin. This C-chain is formed by the self-glucosylation of the glycogenin, forming a short primer chain. From the C-chain grows out B-chains, and from B-chains branch out B- and A-chains. The B-chains have on average 2 branch points, while the A-chains are terminal, thus unbranched. On average, each chain has length 12, tightly constrained to be between 11 and 15. All A-chains reach the spherical surface of the glycogen.[23][24]
Glycogen in muscle, liver, and fat cells is stored in a hydrated form, composed of three or four parts of water per part of glycogen associated with 0.45 millimoles (18 mg) of potassium per gram of glycogen.[5]
Glucose is an osmotic molecule, and can have profound effects on osmotic pressure in high concentrations possibly leading to cell damage or death if stored in the cell without being modified.[3] Glycogen is a non-osmotic molecule, so it can be used as a solution to storing glucose in the cell without disrupting osmotic pressure.[3]
Functions
[edit]Liver
[edit]As a meal containing carbohydrates or protein is eaten and digested, blood glucose levels rise, and the pancreas secretes insulin. Blood glucose from the portal vein enters liver cells (hepatocytes). Insulin acts on the hepatocytes to stimulate the action of several enzymes, including glycogen synthase. Glucose molecules are added to the chains of glycogen as long as both insulin and glucose remain plentiful. In this postprandial or "fed" state, the liver takes in more glucose from the blood than it releases.
After a meal has been digested and glucose levels begin to fall, insulin secretion is reduced, and glycogen synthesis stops. When it is needed for energy, glycogen is broken down and converted again to glucose. Glycogen phosphorylase is the primary enzyme of glycogen breakdown. For the next 8–12 hours, glucose derived from liver glycogen is the primary source of blood glucose used by the rest of the body for fuel.
Glucagon, another hormone produced by the pancreas, in many respects serves as a countersignal to insulin. In response to insulin levels being below normal (when blood levels of glucose begin to fall below the normal range), glucagon is secreted in increasing amounts and stimulates both glycogenolysis (the breakdown of glycogen) and gluconeogenesis (the production of glucose from other sources).
Muscle
[edit]
Muscle glycogen appears to function as a reserve of quickly available phosphorylated glucose, in the form of glucose-1-phosphate, for muscle cells. Glycogen contained within skeletal muscle cells are primarily in the form of β particles.[25] Other cells that contain small amounts use it locally as well. As muscle cells lack glucose-6-phosphatase, which is required to pass glucose into the blood, the glycogen they store is available solely for internal use and is not shared with other cells. This is in contrast to liver cells, which, on demand, readily do break down their stored glycogen into glucose and send it through the blood stream as fuel for other organs.[26]
Skeletal muscle needs ATP (provides energy) for muscle contraction and relaxation, in what is known as the sliding filament theory. Skeletal muscle relies predominantly on glycogenolysis for the first few minutes as it transitions from rest to activity, as well as throughout high-intensity aerobic activity and all anaerobic activity.[27] During anaerobic activity, such as weightlifting and isometric exercise, the phosphagen system (ATP-PCr) and muscle glycogen are the only substrates used as they do not require oxygen nor blood flow.[27]
Different bioenergetic systems produce ATP at different speeds, with ATP produced from muscle glycogen being much faster than fatty acid oxidation.[28] The level of exercise intensity determines how much of which substrate (fuel) is used for ATP synthesis also. Muscle glycogen can supply a much higher rate of substrate for ATP synthesis than blood glucose. During maximum intensity exercise, muscle glycogen can supply 40 mmol glucose/kg wet weight/minute,[29] whereas blood glucose can supply 4 - 5 mmol.[30][4] Due to its high supply rate and quick ATP synthesis, during high-intensity aerobic activity (such as brisk walking, jogging, or running), the higher the exercise intensity, the more the muscle cell produces ATP from muscle glycogen.[31] This reliance on muscle glycogen is not only to provide the muscle with enough ATP during high-intensity exercise, but also to maintain blood glucose homeostasis (that is, to not become hypoglycaemic by the muscles needing to extract far more glucose from the blood than the liver can provide).[30] A deficit of muscle glycogen leads to muscle fatigue known as "hitting the wall" or "the bonk" (see below under glycogen depletion).
Structure Type
[edit]In 1999, Meléndez et al claimed that the structure of glycogen is optimal under a particular metabolic constraint model, where the structure was suggested to be "fractal" in nature.[32] However, research by Besford et al[33] used small angle X-ray scattering experiments accompanied by branching theory models to show that glycogen is a randomly hyperbranched polymer nanoparticle. Glycogen is not fractal in nature. This has been subsequently verified by others who have performed Monte Carlo simulations of glycogen particle growth, and shown that the molecular density reaches a maximum near the centre of the nanoparticle structure, not at the periphery (contradicting a fractal structure that would have greater density at the periphery).[34]
History
[edit]Glycogen was discovered by Claude Bernard. His experiments showed that the liver contained a substance that could give rise to reducing sugar by the action of a "ferment" in the liver. By 1857, he described the isolation of a substance he called "la matière glycogène", or "sugar-forming substance". Soon after the discovery of glycogen in the liver, M.A. Sanson found that muscular tissue also contains glycogen. The empirical formula for glycogen of (C
6H
10O
5)n was established by August Kekulé in 1858.[35]
Sanson, M. A. "Note sur la formation physiologique du sucre dans l’economie animale." Comptes rendus des seances de l’Academie des Sciences 44 (1857): 1323-5.
Metabolism
[edit]Synthesis
[edit]Glycogen synthesis is, unlike its breakdown, endergonic—it requires the input of energy. Energy for glycogen synthesis comes from uridine triphosphate (UTP), which reacts with glucose-1-phosphate, forming UDP-glucose, in a reaction catalysed by UTP—glucose-1-phosphate uridylyltransferase. Glycogen is synthesized from monomers of UDP-glucose initially by the protein glycogenin, which has two tyrosine anchors for the reducing end of glycogen, since glycogenin is a homodimer. After about eight glucose molecules have been added to a tyrosine residue, the enzyme glycogen synthase progressively lengthens the glycogen chain using UDP-glucose, adding α(1→4)-bonded glucose to the nonreducing end of the glycogen chain.[36]
The glycogen branching enzyme catalyzes the transfer of a terminal fragment of six or seven glucose residues from a nonreducing end to the C-6 hydroxyl group of a glucose residue deeper into the interior of the glycogen molecule. The branching enzyme can act upon only a branch having at least 11 residues, and the enzyme may transfer to the same glucose chain or adjacent glucose chains.
Breakdown
[edit]Glycogen is cleaved from the nonreducing ends of the chain by the enzyme glycogen phosphorylase to produce monomers of glucose-1-phosphate:

In vivo, phosphorolysis proceeds in the direction of glycogen breakdown because the ratio of phosphate and glucose-1-phosphate is usually greater than 100.[37] Glucose-1-phosphate is then converted to glucose 6 phosphate (G6P) by phosphoglucomutase. A special debranching enzyme is needed to remove the α(1→6) branches in branched glycogen and reshape the chain into a linear polymer. The G6P monomers produced have three possible fates:
- G6P can continue on the glycolysis pathway and be used as fuel.
- G6P can enter the pentose phosphate pathway via the enzyme glucose-6-phosphate dehydrogenase to produce NADPH and 5 carbon sugars.
- In the liver and kidney, G6P can be dephosphorylated back to glucose by the enzyme glucose 6-phosphatase. This is the final step in the gluconeogenesis pathway.
Clinical relevance
[edit]Disorders of glycogen metabolism
[edit]The most common disease in which glycogen metabolism becomes abnormal is diabetes, in which, because of abnormal amounts of insulin, liver glycogen can be abnormally accumulated or depleted. Restoration of normal glucose metabolism usually normalizes glycogen metabolism, as well.
In hypoglycemia caused by excessive insulin, liver glycogen levels are high, but the high insulin levels prevent the glycogenolysis necessary to maintain normal blood sugar levels. Glucagon is a common treatment for this type of hypoglycemia.
Various inborn errors of carbohydrate metabolism are caused by deficiencies of enzymes or transport proteins necessary for glycogen synthesis or breakdown. These are collectively referred to as glycogen storage diseases.
Glycogen depletion and endurance exercise
[edit]Long-distance athletes, such as marathon runners, cross-country skiers, and cyclists, often experience glycogen depletion, where almost all of the athlete's glycogen stores are depleted after long periods of exertion without sufficient carbohydrate consumption. This phenomenon is referred to as "hitting the wall" in running and "bonking" in cycling.
Glycogen depletion can be forestalled in three possible ways:
- First, during exercise, carbohydrates with the highest possible rate of conversion to blood glucose (high glycemic index) are ingested continuously. The best possible outcome of this strategy replaces about 35% of glucose consumed at heart rates above about 80% of maximum.
- Second, through endurance training adaptations and specialized regimens (e.g. fasting, low-intensity endurance training), the body can condition type I muscle fibers to improve both fuel use efficiency and workload capacity to increase the percentage of fatty acids used as fuel,[38][39] sparing carbohydrate use from all sources.
- Third, by consuming large quantities of carbohydrates after depleting glycogen stores as a result of exercise or diet, the body can increase storage capacity of intramuscular glycogen stores.[13][40][41][42] This process is known as carbohydrate loading. In general, glycemic index of carbohydrate source does not matter since muscular insulin sensitivity is increased as a result of temporary glycogen depletion.[43][44]
When athletes ingest both carbohydrate and caffeine following exhaustive exercise, their glycogen stores tend to be replenished more rapidly;[45][46] however, the minimum dose of caffeine at which there is a clinically significant effect on glycogen repletion has not been established.[46]
Nanomedicine
[edit]Recently, Glycogen Nanoparticles have been investigated as potential drug delivery systems.[47]
See also
[edit]References
[edit]- ^ McArdle, William D.; Katch, Frank I.; Katch, Victor L. (2006). Exercise physiology: Energy, nutrition, and human performance (6th ed.). Lippincott Williams & Wilkins. p. 12. ISBN 978-0-7817-4990-9.
- ^ Sadava, David E.; Purves, William K.; Hillis, David M.; Orians, Gordon H.; Heller, H. Craig (2011). Life (9th ed.). W. H. Freeman. ISBN 9781429254311.
- ^ a b c Berg JM, Tymoczko JL, Gatto GJ, Stryer L (8 April 2015). Biochemistry (8th ed.). New York: W. H. Freeman. ISBN 9781464126109. OCLC 913469736.
- ^ a b c d e f g h i j Wasserman DH (January 2009). "Four grams of glucose". American Journal of Physiology. Endocrinology and Metabolism. 296 (1): E11–21. doi:10.1152/ajpendo.90563.2008. PMC 2636990. PMID 18840763.
Four grams of glucose circulates in the blood of a person weighing 70 kg. This glucose is critical for normal function in many cell types. In accordance with the importance of these 4 g of glucose, a sophisticated control system is in place to maintain blood glucose constant. Our focus has been on the mechanisms by which the flux of glucose from liver to blood and from blood to skeletal muscle is regulated. ... The brain consumes ~60% of the blood glucose used in the sedentary, fasted person. ... The amount of glucose in the blood is preserved at the expense of glycogen reservoirs (Fig. 2). In postabsorptive humans, there are ~100 g of glycogen in the liver and ~400 g of glycogen in muscle. Carbohydrate oxidation by the working muscle can go up by ~10 fold with exercise, and yet after 1 h, blood glucose is maintained at ~4 g.
- ^ a b Kreitzman SN, Coxon AY, Szaz KF (July 1992). "Glycogen storage: Illusions of easy weight loss, excessive weight regain, and distortions in estimates of body composition". The American Journal of Clinical Nutrition. 56 (1, Suppl): 292s–293s. doi:10.1093/ajcn/56.1.292S. PMID 1615908.
- ^ Guyton, Arthur C.; Hall, John Edward (2011). Guyton and Hall Textbook of Medical Physiology. New York, New York: Saunders/Elsevier. ISBN 978-5-98657-013-6.
- ^ Moses SW, Bashan N, Gutman A (December 1972). "Glycogen metabolism in the normal red blood cell". Blood. 40 (6): 836–843. doi:10.1182/blood.V40.6.836.836. PMID 5083874.
- ^ Ingermann RL, Virgin GL (1987). "Glycogen content and release of glucose from red blood cells of the sipunculan worm themiste dyscrita" (PDF). J Exp Biol. 129: 141–149. doi:10.1242/jeb.129.1.141.
- ^ Miwa I, Suzuki S (November 2002). "An improved quantitative assay of glycogen in erythrocytes". Annals of Clinical Biochemistry. 39 (Pt 6): 612–13. doi:10.1258/000456302760413432. PMID 12564847.
- ^ Murray, Bob (April 2018). "Fundamentals of glycogen metabolism for coaches and athletes". Nutrition Reviews. 76 (4): 243–259. doi:10.1093/nutrit/nuy001. PMC 6019055. PMID 29444266.
- ^ Oe Y, Baba O, Ashida H, Nakamura KC, Hirase H (June 2016). "Glycogen distribution in the microwave-fixed mouse brain reveals heterogeneous astrocytic patterns". Glia. 64 (9): 1532–1545. doi:10.1002/glia.23020. PMC 5094520. PMID 27353480.
- ^ Campbell, Neil A.; Williamson, Brad; Heyden, Robin J. (2006). Biology: Exploring Life. Boston, MA: Pearson Prentice Hall. ISBN 978-0-13-250882-7.
- ^ a b Jensen, Rasmus; Ørtenblad, Niels; Stausholm, Marie-Louise Holleufer; Skjærbæk, Mette Carina; Larsen, Daniel Nykvist; Hansen, Mette; Holmberg, Hans-Christer; Plomgaard, Peter; Nielsen, Joachim (1 October 2020). "Heterogeneity in subcellular muscle glycogen utilisation during exercise impacts endurance capacity in men". The Journal of Physiology. 598 (19): 4271–4292. doi:10.1113/JP280247. ISSN 1469-7793. PMID 32686845. S2CID 220653138.
- ^ a b Jensen, Rasmus; Ørtenblad, Niels; Stausholm, Marie-Louise H.; Skjærbæk, Mette C.; Larsen, Daniel N.; Hansen, Mette; Holmberg, Hans-Christer; Plomgaard, Peter; Nielsen, Joachim (1 May 2021). "Glycogen supercompensation is due to increased number, not size, of glycogen particles in human skeletal muscle". Experimental Physiology. 106 (5): 1272–1284. doi:10.1113/EP089317. ISSN 0958-0670. PMID 33675088. S2CID 232131416.
- ^ Bergström, Jonas; Hermansen, Lars; Hultman, Eric; Saltin, Bengt (October 1967). "Diet, Muscle Glycogen and Physical Performance". Acta Physiologica Scandinavica. 71 (2–3): 140–150. doi:10.1111/j.1748-1716.1967.tb03720.x. ISSN 1365-201X. PMID 5584523.
- ^ Marchand, I.; Chorneyko, K.; Tarnopolsky, M.; Hamilton, S.; Shearer, J.; Potvin, J.; Graham, T. E. (1 November 2002). "Quantification of subcellular glycogen in resting human muscle: granule size, number, and location". Journal of Applied Physiology. 93 (5): 1598–1607. doi:10.1152/japplphysiol.00585.2001. ISSN 8750-7587. PMID 12381743.
- ^ Ryley, J.F. (March 1955). "Studies on the metabolism of the protozoa. 5: Metabolism of the parasitic flagellate Trichomonas foetus". The Biochemical Journal. 59 (3): 361–369. doi:10.1042/bj0590361. PMC 1216250. PMID 14363101.
- ^ Benchimol, Marlene; Elias, Cezar Antonio; de Souza, Wanderley (December 1982). "Tritrichomonas foetus: Ultrastructural localization of calcium in the plasma membrane and in the hydrogenosome". Experimental Parasitology. 54 (3): 277–284. doi:10.1016/0014-4894(82)90036-4. ISSN 0014-4894. PMID 7151939.
- ^ Mielewczik, Michael; Mehlhorn, Heinz; al Quraishy, Saleh; Grabensteiner, E.; Hess, M. (1 September 2008). "Transmission electron microscopic studies of stages of histomonas meleagridis from clonal cultures". Parasitology Research. 103 (4): 745–750. doi:10.1007/s00436-008-1009-1. ISSN 0932-0113. PMID 18626664. S2CID 2331300.
- ^ Manners, David J. (1991). "Recent developments in our understanding of glycogen structure". Carbohydrate Polymers. 16 (1): 37–82. doi:10.1016/0144-8617(91)90071-J. ISSN 0144-8617.
- ^ Ronner, Peter (2018). Netter's Essentials Biochemistry. USA: Elsevier. p. 254. ISBN 978-1-929007-63-9.
- ^ Berg, Jeremy Mark; Tymoczko, John L.; Stryer, Lubert (2012). Biochemistry (7th ed.). W. H. Freeman. p. 338. ISBN 978-1429203142.
- ^ Gunja-Smith, Zeenat; Marshall, J.J.; Mercier, Christiane; Smith, E.E.; Whelan, W.J. (28 December 1970). "A revision of the Meyer-Bernfeld model of glycogen and amylopectin". FEBS Letters. 12 (2): 101–104. doi:10.1016/0014-5793(70)80573-7. ISSN 0014-5793. PMID 11945551. S2CID 34722785.
- ^ Roach, Peter J.; Depaoli-Roach, Anna A.; Hurley, Thomas D.; Tagliabracci, Vincent S. (16 January 2012). "Glycogen and its metabolism: some new developments and old themes". Biochemical Journal. 441 (3): 763–787. doi:10.1042/BJ20111416. ISSN 0264-6021. PMC 4945249. PMID 22248338.
- ^ Liu, Q. H.; Wang, Z. Y.; Tang, J. W.; Mou, J. Y.; Ma, Z. W.; Deng, B.; Liu, Z.; Wang, L. (2022). "BrowZine". Carbohydrate Polymers. 295. doi:10.1016/j.carbpol.2022.119710. PMID 35989025. S2CID 249489284. Retrieved 12 May 2023.
- ^ "Glycogen Biosynthesis; Glycogen Breakdown". oregonstate.edu. Archived from the original on 12 May 2021. Retrieved 28 February 2018.
- ^ a b Lucia, Alejandro; Martinuzzi, Andrea; Nogales-Gadea, Gisela; Quinlivan, Ros; Reason, Stacey; International Association for Muscle Glycogen Storage Disease study group (December 2021). "Clinical practice guidelines for glycogen storage disease V & VII (McArdle disease and Tarui disease) from an international study group". Neuromuscular Disorders. 31 (12): 1296–1310. doi:10.1016/j.nmd.2021.10.006. ISSN 1873-2364. PMID 34848128. S2CID 240123241.
- ^ "Hormonal Regulation of Energy Metabolism - Berne and Levy Physiology, 6th ed (2008)". doctorlib.info. Retrieved 21 October 2023.
- ^ Murray, Bob; Rosenbloom, Christine (1 April 2018). "Fundamentals of glycogen metabolism for coaches and athletes". Nutrition Reviews. 76 (4): 243–259. doi:10.1093/nutrit/nuy001. ISSN 1753-4887. PMC 6019055. PMID 29444266.
- ^ a b Brooks, George A. (January 2020). "The Precious Few Grams of Glucose During Exercise". International Journal of Molecular Sciences. 21 (16): 5733. doi:10.3390/ijms21165733. ISSN 1422-0067. PMC 7461129. PMID 32785124.
- ^ van Loon, L. J.; Greenhaff, P. L.; Constantin-Teodosiu, D.; Saris, W. H.; Wagenmakers, A. J. (1 October 2001). "The effects of increasing exercise intensity on muscle fuel utilisation in humans". The Journal of Physiology. 536 (Pt 1): 295–304. doi:10.1111/j.1469-7793.2001.00295.x. ISSN 0022-3751. PMC 2278845. PMID 11579177.
- ^ Ruth Melendez; Enrique Melendez-Hevia; Enric I. Canela (September 1999). "The Fractal Structure of Glycogen: A Clever Solution to Optimize Cell Metabolism". Biophysical Journal. 77 (3). 1327. doi:10.1016/S0006-3495(99)76982-1. hdl:2445/122234.
- ^ Quinn A. Besford; Xiao-Yi Zeng; Ji-Ming Ye; Angus Gray-Weale (31 October 2015) [31 October 2015]. "Liver glycogen in type 2 diabetic mice is randomly branched as enlarged aggregates with blunted glucose release". Glycoconjugate Journal. 33. 41-51. doi:10.1007/s10719-015-9631-5. hdl:11343/282927.
- ^ Peng Zhang; Sharif S. Nada; Xinle Tan; Bin Deng; Mitchell A. Sullivan; Robert G. Gilbert (8 May 2018) [8 May 2018]. "Exploring glycogen biosynthesis through Monte Carlo simulation". International Journal of Biological Macromolecules. 116. 264-271. doi:10.1016/j.ijbiomac.2018.05.027.
- ^ Young, F.G. (22 June 1957). "Claude Bernard and the discovery of glycogen". British Medical Journal. 1 (5033): 1431–1437. doi:10.1136/bmj.1.5033.1431. JSTOR 25382898. PMC 1973429. PMID 13436813.
- ^ Nelson, D. (2013). Lehninger Principles of Biochemistry (6th ed.). W.H. Freeman and Company. p. 618.
- ^ Stryer, L. (1988). Biochemistry (3rd ed.). Freeman. p. 451.
- ^ "Methods of endurance training, Part 1". 30 October 2009. Archived from the original on 22 July 2018. Retrieved 1 August 2013.
- ^ "Steady state vs. tempo training and fat loss". 2 June 2008. Archived from the original on 5 September 2017. Retrieved 1 August 2013.
- ^ McDonald, Lyle (25 July 2012). "Research review: An in-depth look into carbing up on the cyclical ketogenic diet". Archived from the original on 11 November 2020. Retrieved 19 February 2017.
- ^ McDonald, Lyle (1998). The Ketogenic Diet: A complete guide for the dieter and the practitioner. Lyle McDonald.
- ^ Costill DL, Bowers R, Branam G, Sparks K (December 1971). "Muscle glycogen utilization during prolonged exercise on successive days". J Appl Physiol. 31 (6): 834–838. doi:10.1152/jappl.1971.31.6.834. PMID 5123660.
- ^ Zorzano A, Balon TW, Goodman MN, Ruderman NB (December 1986). "Glycogen depletion and increased insulin sensitivity and responsiveness in muscle after exercise". Am. J. Physiol. 251 (6, Part 1): E664–E669. doi:10.1152/ajpendo.1986.251.6.E664. PMID 3538900.
- ^ McDonald, Lyle (2003). The Ultimate Diet 2.0. Lyle McDonald.
- ^ Pedersen, D.J.; Lessard, S.J.; Coffey, V.G.; et al. (July 2008). "High rates of muscle glycogen resynthesis after exhaustive exercise when carbohydrate is coingested with caffeine". Journal of Applied Physiology. 105 (1): 7–13. doi:10.1152/japplphysiol.01121.2007. PMID 18467543.
- ^ a b Beelen, M.; Burke, L.M.; Gibala, M.J.; van Loon, L.J.C. (December 2010). "Nutritional strategies to promote post-exercise recovery". International Journal of Sport Nutrition and Exercise Metabolism. 20 (6): 515–532. doi:10.1123/ijsnem.20.6.515. PMID 21116024. S2CID 13748227.
- ^ Quinn A. Besford; Francesca Cavalieri; Frank Caruso (7 May 2020) [16 October 2019]. "Glycogen as a Building Block for Advanced Biological Materials". Advanced Materials. 32 (18). 1904625. Bibcode:2020AdM....3204625B. doi:10.1002/adma.201904625. hdl:11343/230737. PMID 31617264. S2CID 204738366.
External links
[edit]- "Glycogen storage disease". McArdle's Diseases.
- Glycogen at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
Types of carbohydrates | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| General | |||||||||||||||
| Geometry | |||||||||||||||
| Monosaccharides |
| ||||||||||||||
| Multiple |
| ||||||||||||||